健康な日本人の腸内細菌叢の特徴解明、約500万の遺伝子を発見 日本人は生体に有益な機能が外国よりも多く平均寿命の高さや低肥満率等との関連も示唆
早稲田大学理工学術院先進理工学研究科の服部正平(はっとりまさひら)教授と東京大学大学院新領域創成科学研究科の西嶋傑(にしじますぐる)博士課程学生らを中心とする共同研究グループ(#後述参照)は、日本人を含めた12カ国のヒト腸内細菌叢データの比較解析を行い、腸内細菌叢の菌種組成が国ごとで大きく異なることや日本人の腸内細菌叢の特徴を明らかにしました。
共同研究グループは、106名の日本人の腸内細菌叢※1の大規模なメタゲノム解析※2を行い、①日本人腸内細菌叢に約500万の遺伝子を発見し、外国も合わせて少なくとも1,200万の遺伝子をもつこと、②同じ国の被験者間の細菌叢の類似性が他国の被験者間の類似性よりも有意に高い、すなわち、国ごとに特徴的な細菌叢が形成されること等を明らかにしました。さらに、日本人データと欧・米・中国等の外国11カ国データとの比較解析から、日本人腸内細菌叢は、①ビフィズス菌やブラウチア等が優勢し、古細菌が少ない、②炭水化物やアミノ酸代謝の機能が豊富である一方で、細胞運動性や複製・修復機能が少ない、③他の11カ国ではおもにメタン生成に消費される水素が日本人ではおもに酢酸生成に消費される等の違いや特徴が明らかとなりました。このほか、④海苔やワカメ(の多糖類)を分解する酵素遺伝子が、約90%の日本人に保有されるのに対して、他の11カ国では〜15%となり、本酵素が日本人集団に特徴的に広く分布していることも明らかとなりました。以上のような日本人腸内細菌叢の特徴には、生体に有益な機能が外国よりも多く含まれ、その総合的な有益性は日本人の世界一の平均寿命や低い肥満率等と関連することが示唆されました。
今回の成果は、ヒト腸内細菌叢の集団レベルでの多様性と日本人の腸内細菌叢の特徴を世界で初めて明らかにしたもので、今後、腸内細菌叢が関与する病気の治療や予防、健康増進に役立つ生活習慣の改善等への応用が期待されます。
本研究成果は、科学雑誌『DNA Research』(3月6日online版)に掲載されました。
論文名:The gut microbiome of healthy Japanese and its microbial and functional uniqueness
# 共同研究グループ
早稲田大学理工学術院先進理工学研究科(服部 正平)
東京大学大学院新領域創成科学研究科(西嶋 傑、服部 正平、須田 亙、大島 健志朗*)
慶應義塾大学医学部(須田 亙)
理化学研究所統合生命医科学研究センター(金 錫元)
岡山大学農学部(森田 英利)
豊橋技術科学大学環境・生命工学系(広瀬 侑)
*科学技術振興機構CREST「生体恒常性維持・変容破綻機構のネットワーク的理解に基づく最適医療実現のため技術創出」
1. 背 景
メタゲノム技術の著しい進歩により、これまで困難であったヒトの腸内細菌叢の全体像が解明されつつあります。近年では、欧米、中国、南米やアフリカの原住民等、さまざまな国・地域から疾患患者を含めて〜2,000名の腸内細菌叢のメタゲノム配列データが蓄積されています。このような国や食習慣、生活習慣が異なったヒト集団の腸内細菌叢はそれらの違いを反映してお互いに異なることが示唆されているのですが、その具体的な相違は断片的にしか知られていません。とくに、食習慣や生活習慣が欧米諸国と異なり、また、先進国の中できわめて低いBMIと世界一の平均寿命をもつ日本人の腸内細菌叢の解明は魅力的です。しかし、これまでに発表された日本人腸内細菌叢のメタゲノム配列データ量は少なく、その全体像や特性を知るには不十分でした。また、一方で、各種病態の腸内細菌叢の研究から、腸内細菌叢の変容(ディスバイオシス※3)が肥満、糖尿病、炎症性腸疾患、自閉症等の種々の疾患の発症と関連することが明らかとなりました。このことは腸内環境が宿主のさまざまな生理状態に密接に関係することを示唆します。このディスバイオシスの評価・判断には基準となる健常者コントロール群の腸内細菌叢データが必要となります。しかし、上述したように、日本人健常者のデータは不足しており、日本人腸内細菌叢のディスバイオシスを十分に評価することができません。また、もし国間の腸内細菌叢が大きく異なれば、外国人データを基準にして日本人腸内細菌叢を正しく評価できません。以上のことから、日本人の低いBMIや長寿と腸内環境・腸内細菌叢との関係性や、日本人の疾患における腸内細菌叢の影響を知るために、まとまった大量の健常日本人の腸内細菌叢メタゲノム配列データを収集・解析することが求められていました。
2.研究手法と成果
日本人腸内細菌叢のメタゲノム解析: 本研究では、106名の大人(19〜60歳;BMI: 22 ± 2.7、女42名、男64名)の糞便から調製した腸内細菌叢のDNAを解析しました。次世代シークエンサー※4を用いて、平均3.4 Gb(ギガ塩基=109塩基)/被験者の細菌DNA配列(メタゲノム配列データ)を収集しました(図1)。このDNA配列を参照ゲノムデータベース※5に相同検索することで、菌種の帰属や菌種組成等の細菌解析、並びに、DNA配列から遺伝子を同定し、同定された遺伝子を機能データベース(KEGG)※6に相同検索することで、それらの機能解析を行いました。同様に、外国人のメタゲノム配列データからそれらの細菌と機能解析を行いました。日本と外国データの比較解析はさまざまな情報解析ツールと統計手法を用いて行いました。 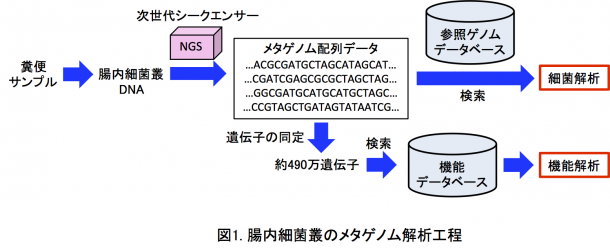
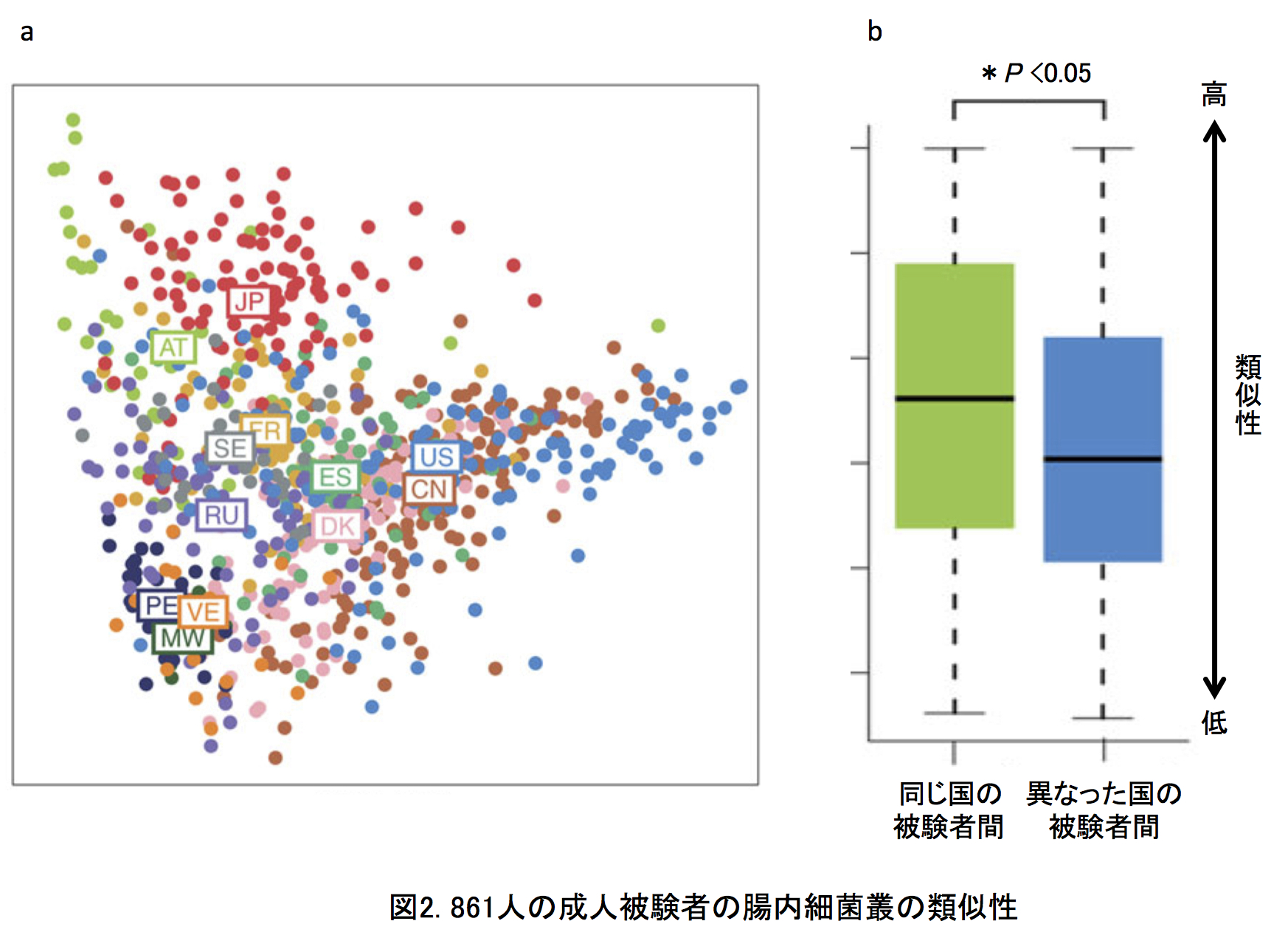
12カ国の腸内細菌叢の比較解析: 本研究では、日本、アメリカ、デンマーク、スペイン、フランス、スウェーデン、オーストリア、ロシア、ペルー、マラウイ、ベネズエラ、中国の12カ国の総数861人の健常者のデータ(BMI30以上の肥満、2型糖尿病、炎症性腸疾患、肝硬変、大腸がんを除いた)を比較しました。図2aに菌種組成の類似性から求めた各被験者の位置を示します(多次元尺度構成法)。細菌叢が似ている被験者は互いに近くに、異なるほど遠くに位置されます。この図から、同じ国の被験者同士が近くに集まって国ごとにクラスターを作る傾向のあることがわかります。図2bには同じ国の被験者間の類似性が他国の被験者間の類似性よりも有意に高いことを示しました。つまり、腸内細菌叢の菌種組成は国ごとに有意に異なっていることが明らかとなりました。この国特異性は、菌種組成からその被験者の出身国を推定したときの平均正答率が87%となったことから、きわめて高いことがわかりました。日本人データの場合、100%の正答率となり、きわめて特異的な腸内細菌叢であることが示唆されました。また、この国間の大きな違いは、個人の腸内細菌叢のディスバイオシスを正しく診断・評価するには、同じ国の健常者データを基準にする必要があることを示しています。つぎに、12カ国の腸内細菌叢について国レベルでの類似性を調べました(図3)。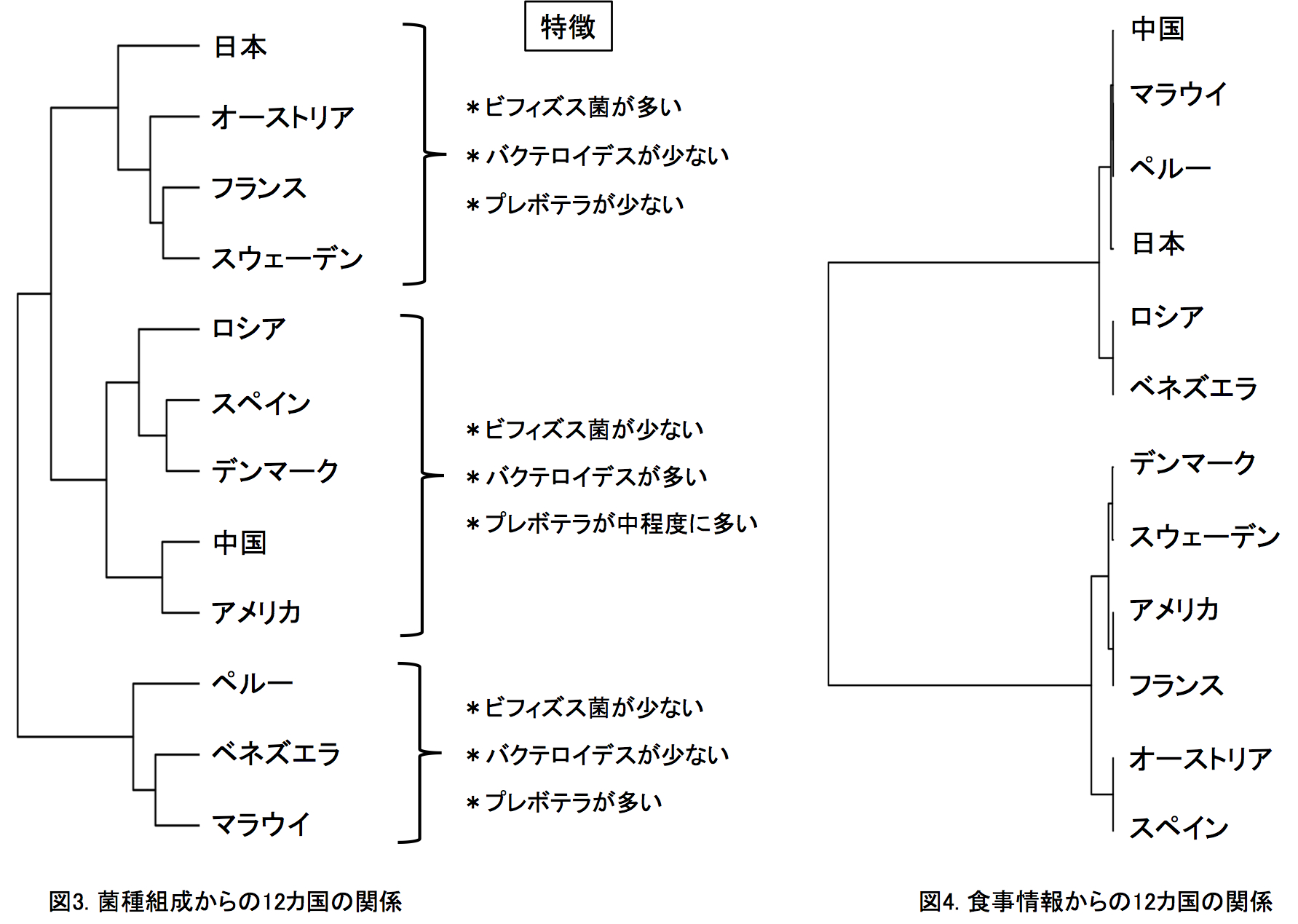 12カ国は、(日本・オーストリア・フランス・スウェーデン)、(アメリカ・中国・デンマーク・スペイン・ロシア)、(マラウイ・ベネズエラ・ペルー)の3つのグループに大きく分かれました。それぞれのグループ間の違いは、概ねビフィズス菌、ブラウチア、バクテロイデス、プレボテラの組成比の違いによるものです。日本は12カ国の中でもっともビフィズス菌とブラウチアが多いという特徴がありました(後述)。興味深いことは、日本が3つの欧州国とグループをつくることや、中国とアメリカがもっとも近い関係にあることです。日本と中国は地理的に近く、同じアジア人であり食習慣も似ているにも関わらずお互いは遠い関係にあり、両国とも欧米諸国と近い関係にありました。一方で、南米のベネズエラ、ペルーとアフリカのマラウイは地理的には遠いが穀物類を主食とする原住民という共通性をもっていました。すなわち、得られた国間の違いを、地理的近さ、人種、食習慣のそれぞれ単独で説明できませんでした。事実、各国の10年間の平均の食事中の栄養素(たんぱく質、脂質、たんぱく質の組成比)を元にした12カ国の関係は、ロシアを除く欧米諸国(たんぱく質が多い食事)とアジア・南米・アフリカ(たんぱく質が少ない食事)の2グループに大きく分かれ、今日の世界の食事様式を的確に示しました(図4)。しかしながら、この食事情報からの12カ国の関係と今回の腸内細菌叢データからの12カ国の関係は必ずしも一致しませんでした。つまり、食事以外に、腸内細菌叢の形成に大きく影響する別のファクターの存在が今回の研究から示唆されました。12カ国の中で日本人腸内細菌叢にもっとも多い5菌種(Bifidobacterium, Blautia, Collinsella, Streptococcus, Unclassified Clostridiales)ともっとも少ない6菌種(Clostridium, Alistipes, Dialister, Butyrivibrio, Unclassified Firmicutes, Methanobrevibacter smithii)を主要な26優勢菌種中に同定しました(図5)。
12カ国は、(日本・オーストリア・フランス・スウェーデン)、(アメリカ・中国・デンマーク・スペイン・ロシア)、(マラウイ・ベネズエラ・ペルー)の3つのグループに大きく分かれました。それぞれのグループ間の違いは、概ねビフィズス菌、ブラウチア、バクテロイデス、プレボテラの組成比の違いによるものです。日本は12カ国の中でもっともビフィズス菌とブラウチアが多いという特徴がありました(後述)。興味深いことは、日本が3つの欧州国とグループをつくることや、中国とアメリカがもっとも近い関係にあることです。日本と中国は地理的に近く、同じアジア人であり食習慣も似ているにも関わらずお互いは遠い関係にあり、両国とも欧米諸国と近い関係にありました。一方で、南米のベネズエラ、ペルーとアフリカのマラウイは地理的には遠いが穀物類を主食とする原住民という共通性をもっていました。すなわち、得られた国間の違いを、地理的近さ、人種、食習慣のそれぞれ単独で説明できませんでした。事実、各国の10年間の平均の食事中の栄養素(たんぱく質、脂質、たんぱく質の組成比)を元にした12カ国の関係は、ロシアを除く欧米諸国(たんぱく質が多い食事)とアジア・南米・アフリカ(たんぱく質が少ない食事)の2グループに大きく分かれ、今日の世界の食事様式を的確に示しました(図4)。しかしながら、この食事情報からの12カ国の関係と今回の腸内細菌叢データからの12カ国の関係は必ずしも一致しませんでした。つまり、食事以外に、腸内細菌叢の形成に大きく影響する別のファクターの存在が今回の研究から示唆されました。12カ国の中で日本人腸内細菌叢にもっとも多い5菌種(Bifidobacterium, Blautia, Collinsella, Streptococcus, Unclassified Clostridiales)ともっとも少ない6菌種(Clostridium, Alistipes, Dialister, Butyrivibrio, Unclassified Firmicutes, Methanobrevibacter smithii)を主要な26優勢菌種中に同定しました(図5)。 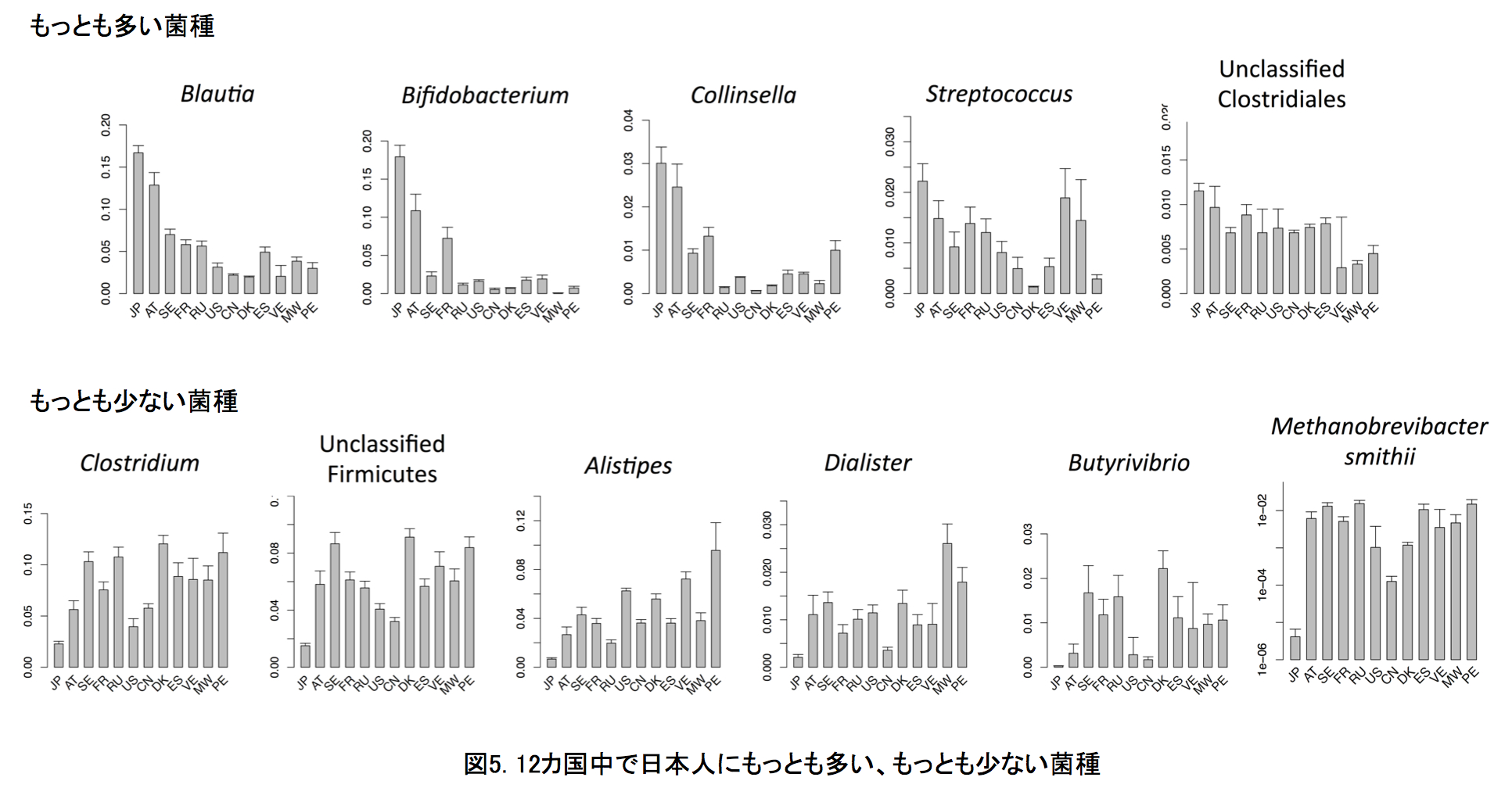
ヒト腸内細菌叢の遺伝子数: 今回の研究で収集された日本人腸内細菌叢に約490万のユニーク遺伝子(いかなる遺伝子に対しても塩基配列の類似度が95%未満となる遺伝子)を情報学的に同定しました。ついで、アメリカ、デンマーク、スペイン、中国の4カ国の腸内細菌叢に同定されている約990万の遺伝子(IGC遺伝子セット)に、今回の日本人の遺伝子をマージすると、ヒト腸内細菌叢のもつユニーク遺伝子の総数が約1,200万となりました。この数はヒト遺伝子の〜2.5万をはるかに多く、ヒト腸内細菌叢が宿主ヒトよりもきわめて多様な遺伝子をもつことがわかります。
 日本人腸内細菌叢の機能の特徴:日本人及び他の11カ国の腸内細菌叢に同定された遺伝子を機能データベース(KEGG)に相同検索することで、12カ国で日本人が他国よりも有意に多いあるいは少ない機能を調べました(図6)。その結果、日本人腸内細菌叢は「炭水化物代謝」、「アミノ酸代謝」、「膜輸送」に関わる機能が外国よりも豊富であること、一方で、鞭毛等の「細胞運動性」やDNA損傷に関わる「複製・修復機能」が外国よりも少ないことがわかりました。また、「エネルギー代謝」と「翻訳」が日本人に少ない理由は、外国に多い古細菌Methanobrevibacter smithiiが日本人にもっとも少ないためです(図5)。日本人の高い炭水化物の取り込みと代謝能は、より多くの短鎖脂肪酸(酢酸や酪酸)、二酸化炭素、水素を生成します(図7)。短鎖脂肪酸はヒト細胞の有用な栄養素のひとつであり、水素は抗酸化剤として働くことが知られており、これらは宿主ヒトに対して有益に作用します。また、細胞運動性の少なさは炎症反応が少ない腸内環境を示唆し、複製・修復の機能の少なさはDNA損傷の少ない腸内環境を示唆します。以上の機能の特徴から、日本人の腸内環境は他の11カ国よりも相対的に健全な状態であることを示唆しています。さらに、炭水化物の代謝で生じる水素は、おもに3つの経路(メタン生成、酢酸生成、硫酸還元)で消費されます(図7)。
日本人腸内細菌叢の機能の特徴:日本人及び他の11カ国の腸内細菌叢に同定された遺伝子を機能データベース(KEGG)に相同検索することで、12カ国で日本人が他国よりも有意に多いあるいは少ない機能を調べました(図6)。その結果、日本人腸内細菌叢は「炭水化物代謝」、「アミノ酸代謝」、「膜輸送」に関わる機能が外国よりも豊富であること、一方で、鞭毛等の「細胞運動性」やDNA損傷に関わる「複製・修復機能」が外国よりも少ないことがわかりました。また、「エネルギー代謝」と「翻訳」が日本人に少ない理由は、外国に多い古細菌Methanobrevibacter smithiiが日本人にもっとも少ないためです(図5)。日本人の高い炭水化物の取り込みと代謝能は、より多くの短鎖脂肪酸(酢酸や酪酸)、二酸化炭素、水素を生成します(図7)。短鎖脂肪酸はヒト細胞の有用な栄養素のひとつであり、水素は抗酸化剤として働くことが知られており、これらは宿主ヒトに対して有益に作用します。また、細胞運動性の少なさは炎症反応が少ない腸内環境を示唆し、複製・修復の機能の少なさはDNA損傷の少ない腸内環境を示唆します。以上の機能の特徴から、日本人の腸内環境は他の11カ国よりも相対的に健全な状態であることを示唆しています。さらに、炭水化物の代謝で生じる水素は、おもに3つの経路(メタン生成、酢酸生成、硫酸還元)で消費されます(図7)。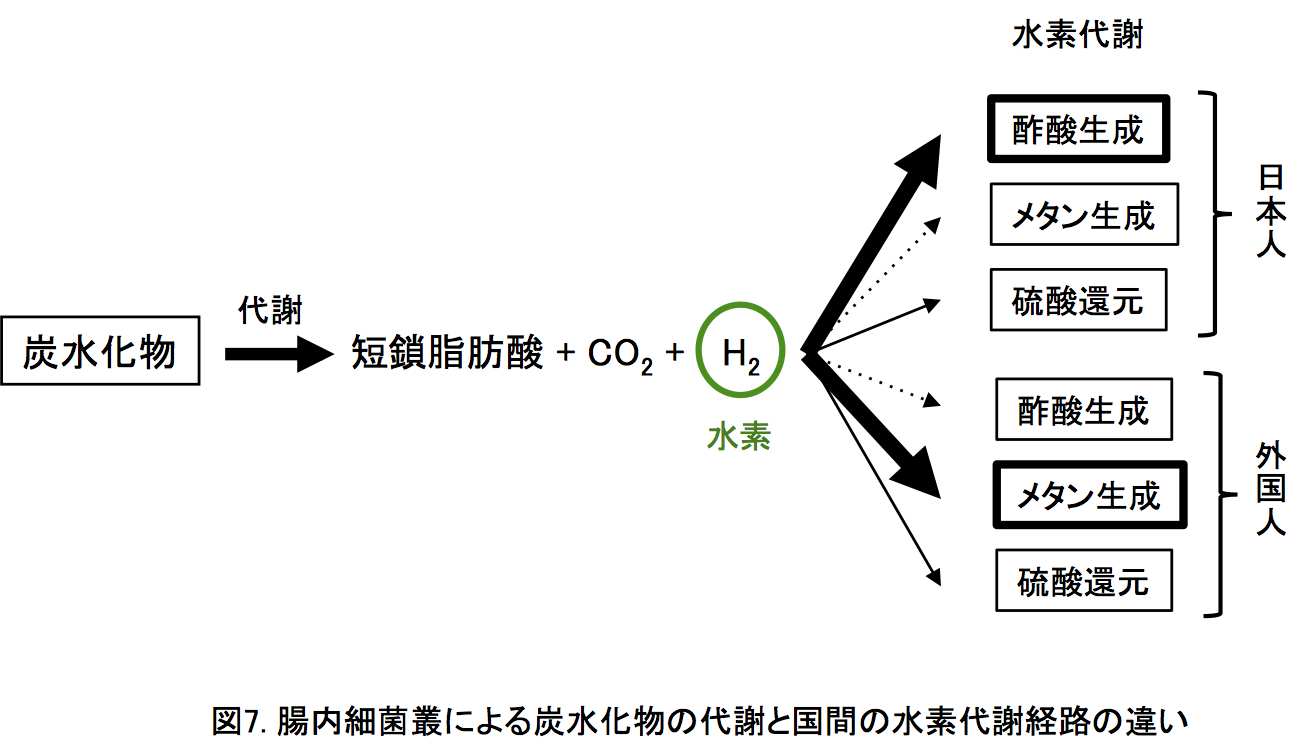 これら水素代謝に関わる遺伝子の量を比較すると、日本人はおもに酢酸生成(ブラウチアがおもに関与)を水素の消費に使い、他の11カ国はおもにメタン生成(Methanobrevibacterがおもに関与)あるいは酢酸生成とメタン生成の両方を水素の消費に使うことが示唆されました。酢酸生成の最終産物は酢酸であり、上述したように、ヒト細胞の有用な栄養素となり、水素代謝も日本人の腸内環境の健全性に寄与します。なお、以前から日本人の呼気に含まれるメタンの量が外国人よりも少ないことが知られていましたが、今回、その理由が腸内細菌による水素代謝の違いであることもわかりました。このほか、水生植物である海苔やワカメ(の多糖類)を分解する酵素(ポルフィラナーゼ)遺伝子が、約90%の日本人に保有されるのに対して、他の11カ国では〜15%となり、本酵素が日本人集団に特徴的に広く分布していることも明らかとなりました。
これら水素代謝に関わる遺伝子の量を比較すると、日本人はおもに酢酸生成(ブラウチアがおもに関与)を水素の消費に使い、他の11カ国はおもにメタン生成(Methanobrevibacterがおもに関与)あるいは酢酸生成とメタン生成の両方を水素の消費に使うことが示唆されました。酢酸生成の最終産物は酢酸であり、上述したように、ヒト細胞の有用な栄養素となり、水素代謝も日本人の腸内環境の健全性に寄与します。なお、以前から日本人の呼気に含まれるメタンの量が外国人よりも少ないことが知られていましたが、今回、その理由が腸内細菌による水素代謝の違いであることもわかりました。このほか、水生植物である海苔やワカメ(の多糖類)を分解する酵素(ポルフィラナーゼ)遺伝子が、約90%の日本人に保有されるのに対して、他の11カ国では〜15%となり、本酵素が日本人集団に特徴的に広く分布していることも明らかとなりました。
3. 今後の展開
本研究では、ヒト腸内細菌叢が国レベルで大きく異なることと日本人の腸内細菌叢の特徴が明らかとなりました。本研究で収集された大量の腸内細菌叢データは、日本人の病態の腸内細菌叢のディスバイオシスを診断する基準データとして、また、腸内細菌叢が関与する病気の予防や治療、健康増進につながる生活習慣の改善等に役立つと期待されます。今後は、今回明らかとなったヒト腸内細菌叢の国特異性や日本人の腸内細菌叢の形成に関与する食事を含めた諸要因を明らかにすることが重要と考えます。
用語解説
- ※1 ヒト腸内細菌叢
ヒトの消化管には種類にして約1,000菌種、菌数にしてヒトの細胞数の約10倍の数百兆個にのぼる腸内細菌の集団(細菌叢)が生息しています。腸内細菌叢はヒトの健康と病気に関係することが知られています。
- ※2 メタゲノム解析
腸内細菌叢を1つの有機体としてとらえてそのDNA情報から全体構造を明らかにする手法。メタゲノム解析には2つの方法があります。細菌のゲノム・遺伝子配列を網羅的に収集・解析する方法(狭義のメタゲノム解析)と、細菌の16SリボソームRNA(rRNA)遺伝子のみを解析する方法(16S解析)です。前者からは細菌情報と遺伝子(=機能)情報の両方が得られ、後者は細菌情報に特化した解析法です。両方法とも、大量の塩基配列データを情報学と統計学を駆使して解析します。
- ※3 腸内細菌叢の変容(ディスバイオシス)
ヒトの腸内細菌叢は肥満や糖尿病などの代謝系の疾患、アレルギーや炎症性腸疾患などの免疫系の疾患、多発性硬化症や自閉症等の神経系の疾患との関係が明らかになっています。これらの疾患では、健康なヒトとは異なった細菌組成や構成菌種からなる腸内細菌叢が形成され、この細菌叢の異常を変容(ディスバイオシス)といいます。この変容がヒトの腸管細胞に作用して、病気を慢性化したり寛解したりすると言われています。
- ※4 次世代シークエンサー(Next Generation Sequencer; NGS)
DNAの塩基配列を自動的に解読する装置をシークエンサーと言います。2008年頃より、従来のシークエンサー(ヒトゲノム計画 1991-2004に使用)の〜100万倍の解読スピードをもつ種々の超高速シークエンサーが実用化され、これらを次世代シークエンサーと言います。
- ※5 参照ゲノムデータベース
腸内細菌も含めた個々のヒト常在菌株のゲノム配列データを格納・管理し、それらを種々の目的に使用できるようにしたコンピュータ上のリソース。これまでに7,000株以上の個別ゲノムデータが登録されています。メタゲノム配列を参照ゲノムデータベースに相同検索することで、各配列データを細菌(ゲノム)に帰属でき、また、菌種組成を定量的に評価することができます。
- ※6 機能データベース(KEGG)
KEGG(Kyoto Encyclopedia of Genes and Genomes)はこれまでに明らかとなった各遺伝子の機能を階層的に分類したリソースデータベースです。本研究では、同定された遺伝子の機能やそれを含む代謝経路の探索に使われました。






