Tension sensor system required for even chromosome segregation
Fri, Sep 27, 2019-
Tags
Solving the puzzle on how SET oncogene causes acute myeloid leukemia
The group led by Yuichiro Asai, a Research Associate, and Professor Yasuhiko Terada at Waseda University, Graduate School of Science and Engineering, found that oncogene SET/TAF1, which was found as a proto-oncogene of acute myeloid leukemia (AML), contributes to the even chromosome segregation as a third tension sensor with Aurora B kinase and PP2A. Additionally, abnormal SET protein disrupts tension sensor system at the centromere, leading to chromosome missegregation and thereby cancer. These findings were obtained through molecular biological techniques and are the discovery that may lead to treatment for leukemia. This research, which was performed collaboratively with the group led by Professor Kyosuke Nagata, a president of Tsukuba University, was published to the online version of Journal of Cell Biology (Rockefeller University Press) in August 2019.
Previous Research
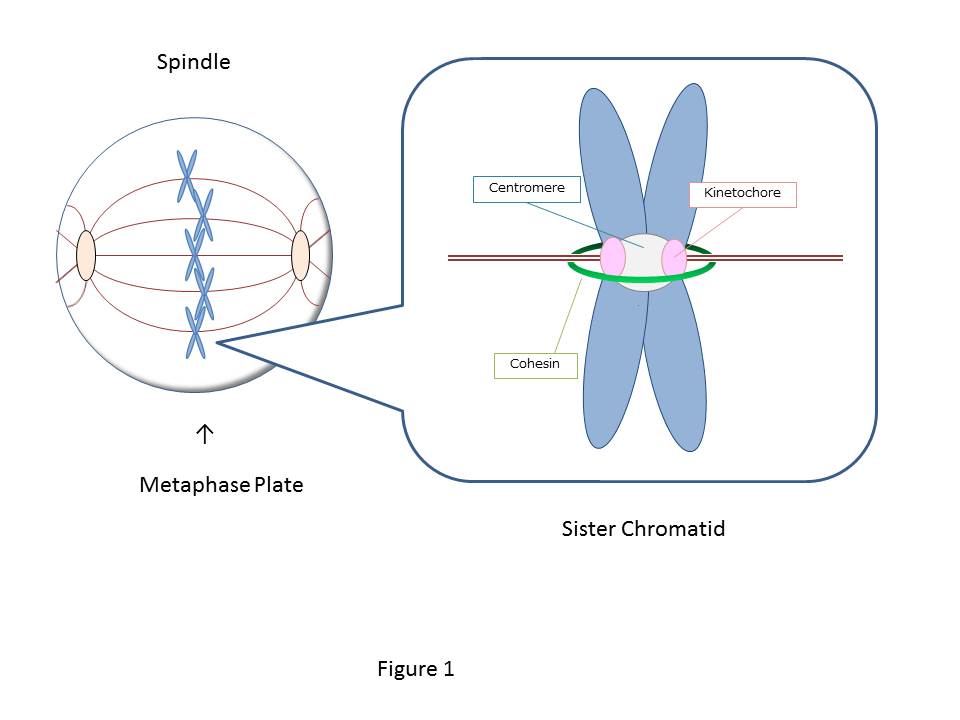
The cell starts from one fertilized egg and divides into about 37 trillion cells, which construct us, human bodies. The 46 human chromosomes are doubled to 92 chromosomes through duplication, and during mitosis, the central region of the duplicated chromosomes called centromere is bundled by a ring-like protein called cohesin (Fig.1). There are two kinetochores (sister kinetochores) in a paired chromosome (sister chromatid), and the microtubule extended from the opposite spindle poles binds to each kinetochore. The pulling force by microtubules acting on the kinetochore aligns the sister chromatids on the equatorial plane (chromosome alignment) and contributes to the even chromosome distribution to the daughter cells. However, even if one chromosome is not aligned, chromosomes are not segregated evenly, and this missegregation causes genetic diseases such as Down syndrome or cancer. To prevent such chromosomal abnormalities, the cell has a system that precisely controls the alignment of all 46 pairs of sister chromatids. The key to this system is thought to be a tension sensor.
The tension sensor locates at each centromeres of 46 sister chromatids, and can accurately detect the tension applied to the sister kinetochores. Under the absence of the tension, the distance between the sister kinetochores is about 0.4 um, but increases to about 1.6um when the microtubule’s pulling force is exerted on the sister kinetochores. Once the tension is applied to all kinetochores in 46 chromosomes, the cohesion is broken down, and the chromosomes are evenly segregated (Fig. 2). But how exactly does the cell distinguishes the difference between the distances on a macro scale: 0.4 um and 1.6 um? The mechanism of this ”ultra-high sensitive tension sensor” was extremely complicated, and its molecular mechanism was not well understood.
In 1996, Terada laboratory searched for new SAC genes in animal cells and succeeded in the world’s first gene cloning of Aurora B/ATM-1, a mitotic kinase that induces mitotic arrest in Saccharomyces cerevisiae, that is an essential protein for chromosome segregation and cytokinesis (Ref. 1,3). Since then, research on Aurora B was conducted worldwide and became clear that Aurora B functions as a part of the tension sensor for even chromosome segregation and cytokinesis. Aurora B, which localizes at the centromere, is activated through autophosphorylation. The activated Aurora B phosphorylates the proteins at the microtubules binding site of the kinetochore, which detaches and corrects the incorrectly attached microtubules (low tension). Once the microtubules are accurately attached, the microtubule’s pulling force gradually increases the distance between the kinetochore and the centromere. When Aurora B at the centromere cannot phosphorylate the distant kinetochore (high tension), the adhesion between the kinetochore and the microtubules strengthens. As a result, Aurora B operates a fundamental role as a tension sensor that corrects the incorrect microtubule attachment to the kinetochore during prometaphase, and gradually stabilizes the attachment during metaphase. On the other hand, the chromosome alignment is known to be controlled by the correlation between Aurora B and PP2A, a phosphatase. However, the regulation mechanism of the tension-dependent Aurora B activity, where it is actively maintained at the low-tension state and weakens as the microtubules stably attach to the kinetochore at the high tension state, has been unexplained for a long period.
In many human cancer cells, the overexpression or abnormal Aurora B was detected, and the overexpression of Aurora B in normal human cells induced chromosomal abnormalities, leading to cancer. Thus in 1998, the abnormality of Aurora B and carcinogenesis were reported to be closely related (Ref. 2). On the other hand, the suppression of the function of Aurora B in cancer cells induces apoptosis (cell death), thereby Aurora B became one of the options as the target proteins for new anticancer drugs (Ref. 4). Based on our findings, many pharmaceutical companies around the world have been developing an anticancer drug that targets Aurora B. AstraZeneca in the United Kingdom have developed a new anticancer drug AZD1152 that targets Aurora B and is currently used in the clinical field as a treatment for AML.
What was revealed in this research
In this research, the SET protein, which is PP2A inhibitor, was revealed to control the kinase activity of Aurora B and PP2A and functions as the third tension sensor during mitosis.
When SET was deleted by RNA interference, the Aurora B activity was dramatically reduced, and the incorrectly attached microtubules at the kinetochore were not corrected, which led to the misalignment of chromosomes. The Aurora B function is activated by autophosphorylation and is inhibited by the dephosphorylation of PP2A, which coexists with Aurora B at centromeres (low tension). Therefore, SET maintains Aurora B activity by inhibiting the PP2A activity at centromeres.
Furthermore, it is intriguing to note that while the chromosomes alignment and the distance between the sister kinetochores increases due to the tension, Aurora B remains at the center of the centromere, whereas PP2A and SET move away from Aurora B towards the kinetochore, and SET subsequently detaches from the kinetochore. This implies that once the chromosomes are aligned, SET detaches from the kinetochore, which promotes the activity of PP2A and weakens the activity of Aurora B, leading to the stable attachment of microtubules to the kinetochore. In summary, when the chromosomes are not aligned, and the tension between the sister kinetochores is low, SET inhibits the activity of PP2A, allowing a high Aurora B activity that corrects the incorrect microtubule attachment. When the chromosomes are aligned, and the tension is applied between the sister kinetochores, SET detaches from the kinetochore and decreases the activity of Aurora B that stabilizes the attachment of microtubules to the kinetochore. SET was found to be controlling this precise mechanism (Fig. 3).
In 1992, SET gene was first discovered in National Cancer Institute (NCI) from SET-CAN fusion gene, which was derived from a patient with AML, and how SET causes the normal cell to become a cancer cell was a mystery. In order to investigate the oncogenic function of SET and SET-CAN, we studied whether centromere localization of SET is essential for chromosomal abnormality. Overexpression of SETβ or SET-CAN wild type, but not a mutants, which are defective in localization at centromeres, caused chromosomal abnormality (aneuploidy). Therefore, SET protein disrupts the function of the central system, the tension sensor, that controls the chromosome alignment, causing chromosome misalignment, and leading to cancer cells. This discovery strongly supports the previous our researches that report the abnormal activity of Aurora B is observed in many human cancer cells, and the overexpression of Aurora B in normal cells induces chromosome misalignment.
Newly developed methods for performing the experiments
To investigate the molecular mechanism of oncogenic SET and SET-CAN, random mutations were introduced into the SET gene using PCR, and a mutant SET gene that cannot be localized in the centromere was established though the yeast-two hybrid screening. It has been revealed that these mutant SET and SET-CAN genes cannot induce chromosomal abnormalities and prevented the cell from becoming cancerous. Therefore, when SET and SET-CAN directly destroy the centromere tension sensor system, the chromosomal abnormality is induced, and the cells become cancerous.
The ripple effect and the impact on the society from this research
In 1996, Aurora B/AIM-1 was discovered in the Terada Laboratory and had been under research for more than 20 years. However, its precise activation control mechanism is still under investigation. In this study, SET, a PP2A inhibitor protein, is localized in the centromere and functions as a third tension sensor by fine-tuning the enzyme activities of Aurora B and PP2A. As these three proteins interact with each other, the 92 replicated chromosomes are distributed evenly to two daughter cells and prevent chromosome abnormalities such as cancer and Down’s syndrome. This discovery revealed the chromosome alignment mechanism, and functions as a baseline for further investigation in the molecular mechanism of cancer malignancy by chromosome missegregation and the development of leukemia. Additionally, anticancer drugs that target SET and Aurora B are under research and development.
Future
Why does the localization of SET, Aurora B, and PP2A change between the centromere and kinetochore when the tension between the sister kinetochores increases? This precise timing and localization are thought to be important for regulating the tension sensor. The mechanism by which SET dissociates from the centromere, PP2A is activated and stabilizes the correct microtubule attachment is still unknown. Solving this mechanism is the key to unravel the mystery of the complicated molecular mechanism of the tension sensor. Is the centromere tension sensor monitoring the attachment of the microtubule to the kinetochore? Or is the distance between the sister kinetochores controlling the attachment? These are unclear. In the future, it is expected that the details of the molecular mechanisms of the tension sensor will be revealed by such research methods.
Figures
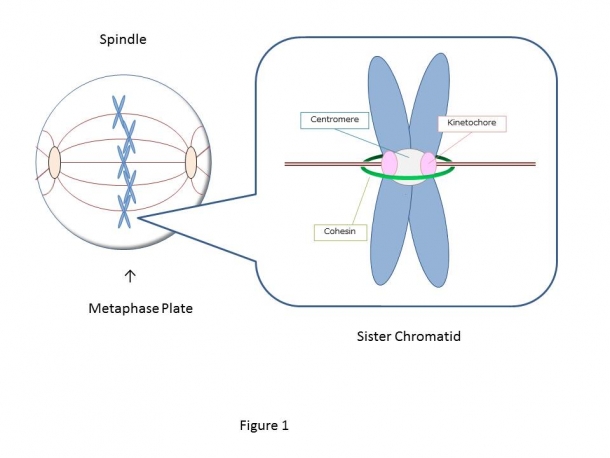
Figure 1: The duplicated chromosomes are paired together by a ring-shaped protein called cohesin (green) and become sister chromatids. There are two kinetochores (pink: sister kinetochores) in a sister chromatids, and when the rope-like microtubules extending from the opposite spindle poles captures each kinetochore, tension is generated, and the chromosomes are aligned on the metaphase plate.
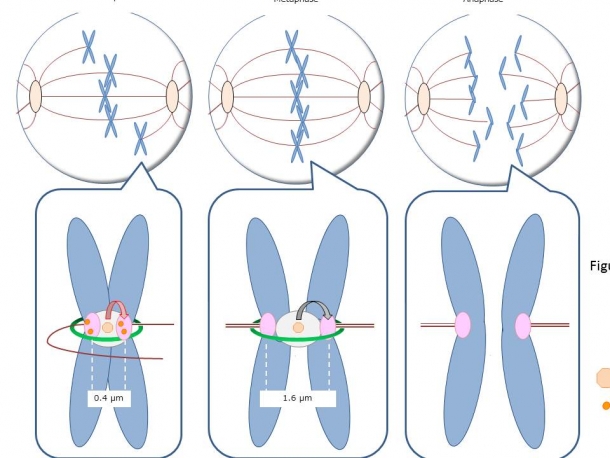
Figure 2: In prometaphase, the sister chromatids are not aligned on the metaphase plate, because the tension applied to the sister kinetochores is uneven. However, as an equal amount of tension is applied to the sister kinetochores, the sister chromatids start to align at the middle, and the cell enters metaphase, where the distance between the sister kinetochores increases from 0.4um to 1.6um. When a high-tension is generated between all 46 sister kinetochores, the checkpoint mechanism called SAC is released, and the cohesin is cleaved, which allows the even chromosome distribution to the daughter cells. Tension sensor is localized at each centromere of 46 sister chromatids, and if one chromosome is not aligned due to the low-tension, SAC prevents the cleavage of cohesin, which arrests the cell at metaphase.
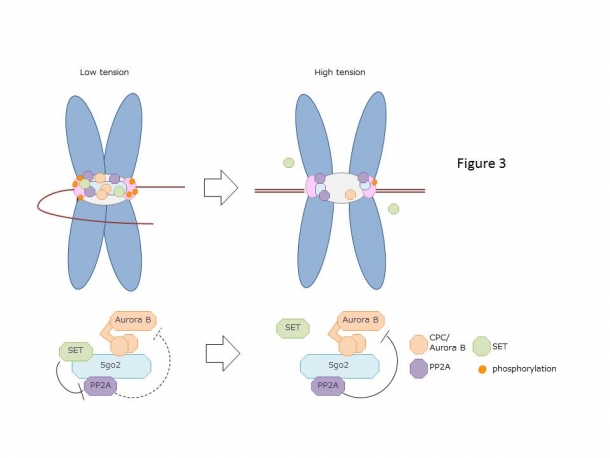
Figure 3: 30 to 40 microtubules can bind to the human kinetochore, but not all microtubules extending from opposite spindle poles are correctly attached at the beginning. When a microtubule is attached to the wrong kinetochore, the tension is low, and the chromosome is not aligned. Aurora B, which is a part of the complex called CPC, corrects the incorrectly attached microtubule by phosphorylating kinetochore proteins. In prometaphase, Aurora B and PP2A co-localize at the centromere with Sgo2 protein as a scaffold. Aurora B is activated through autophosphorylation and is inhibited by the PP2A-mediated dephosphorylation. Since SET interacts directly with PP2A and suppresses its activity, SET has a function of maintaining the Aurora B activity. When all microtubule attachment is corrected, and the distance between the sister kinetochores increase, Aurora B remains at the centromere, whereas PP2A and SET move away from Aurora B towards the kinetochore, and SET subsequently diffuses from the kinetochore to the cytoplasm. Aurora B can no longer phosphorylate the kinetochore proteins, and because SET cannot inhibit the activity of PP2A, PP2A dephosphorylates the phosphorylated kinetochore proteins and stabilizes the correct microtubule attachment. As the tension sensor, Aurora B, PP2A, and SET functions accurately, the balance of phosphorylation at the kinetochore is maintained, which leads to precise chromosome alignment and segregation. However, the abnormal tension sensor leads to chromosome missegregation, thereby aneuploidy, causing cancer and genetic disease such as Down’s syndrome.
Reference
1) AIM-1: a mammalian midbody-associated protein required for cytokinesis. Terada Y, et al., EMBO J. 17:667-76. 1998
2) Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Tatsuka M, et al., Cancer Res.58:4811-6. 1998
3) Role of chromosomal passenger complex in chromosome segregation and cytokinesis. Terada Y. Cell Struct Funct. 26:653-7. 2001
4) Aurora-B/AIM-1 regulates the dynamic behavior of HP1alpha at the G2-M transition. Terada Y. Mol Biol Cell. 17:3232-41. 2006
5) Interaction of Aurora-A and centrosomin at the microtubule-nucleating site in Drosophila and mammalian cells. Terada Y, et al., J Cell Biol. 162:757-63.2003
6) International patent, application number: 9-235371
Title of invention:Cell Cycle Regulator Protein (Aurora-B/AIM-1)
Inventor:CHUGAI PHARMACEUTICAL CO., LTD., Yasuhiko Terada
Application date: December 3rd, 1998
*This press release was translated from the original Japanese version by Mone Yamasaki
Article information
- Paper: “Aurora B kinase activity is regulated by SET/TAF1 on Sgo2 at the inner centromere”
- DOI: 10.1083/jcb.201811060
- Author: Yuichiro Asai, Koh Fukuchi, Yuji Tanno, Saki Koitabashi-Kiyozuka, Tatsuyuki Kiyozuka, Yuko Noda, Rieko Matsumura, Tetsuo Koizumi, Atsushi Watanabe, Kyosuke Nagata, Yoshinori Watanabe, Yasuhiko Terada
- Published in the Journal of Cell Biology on September 16, 2019










