
FUKUNAGA Tsukasa
Assistant Professor
There are still many genes whose functions are unknown
My research field is bioinformatics, and mainly I am developing software for the estimation of gene functions. Genetic information is information that passes the traits of parents, such as black hair and double eyelids, to their children. The complete set of genetic information in an organism, called the genome, is made of a chemical substance called DNA (deoxyribonucleic acid). The parts of the genome that contain the important information needed for carrying out the necessary functions related to life are called genes.
DNA has four types of base: adenine (A), thymine (T), cytosine (C), and guanine (G), and these four types are structurally arranged in the genome. The arrangement of A, T, C, and G (called the letter sequence) represents the genetic information of the genome; there are about three billion letter sequences in the human genome. In recent years, a machine called a sequencer, that automatically reads the letter sequence of the genome, has been developed. The sequencer makes it possible to easily determine the letter sequence of almost all individual organisms, including humans. Research is underway to identify which part of the letter sequence of the genome is a gene, but the function of gene information has not yet been elucidated.
It would take an enormous amount of time to determine the functions of all genes by means of a number of experiments, so it is necessary to involve information science. Therefore, I am conducting research on gene function estimation methods that combine machine learning methods and statistical models.
Estimating function-unknown genes of microorganism
My current research target is microorganisms. Since the amount of information in the genomes of microorganisms is small, the information is easy to read; there are many types of microorganisms; and it is easy to collect samples, so it is possible to obtain a significant amount of data. Another good aspect of microorganisms as a research target is that elucidating the functions of microbial genes will lead to the development of applications for biotechnology and medicine. For example, PCR, which is a term we hear often these days, is a technology that amplifies DNA, using an enzyme called polymerase, which is derived from a microorganism called Thermus aquaticus. In addition, ivermectin, a silver bullet for onchocerciasis, a tropical disease, is made from avermectin produced by a microorganism called streptomyces; so far ivermectin has been administered to more than one billion people.
In conventional microbial research, experiments have been conducted using microorganisms that have been isolated and cultured, to investigate their characteristics and functions. However, it is said that less than 1% of all microorganisms can be isolated and cultured. Nowadays, even in the case of microorganisms, the letter sequences of the genome can be identified, and the gene part in the sequence can be determined, so it is possible to investigate microorganism characteristics and functions from genetic information.
The function estimation method that I am using to estimate the function of function-unknown genes begins with the idea that if, for example, species which have gene B always have gene D as well, gene B and gene D have some relationship (see Fig. 1).

Figure 1. Gene conservation analysis among species
Rows represent species and columns represent genes. 0 indicates that the species does not have the gene, and 1 indicates that it does have. Since all species that have gene B also have gene D, it can be considered that gene B and gene D have some relationship.
A more specific example is shown in Figure 2.
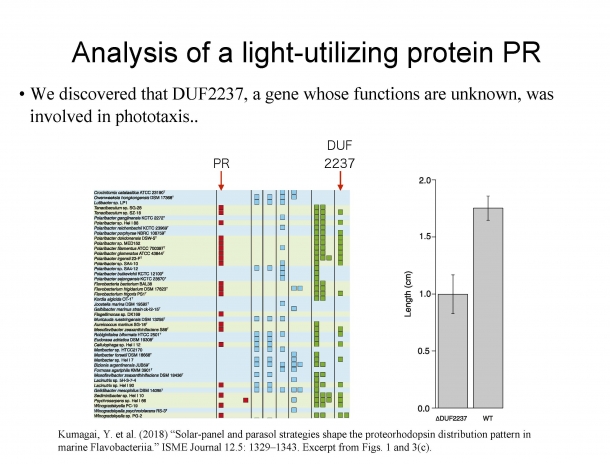
Figure 2. Analysis of a light-utilizing protein PR
In the table on the left, the patterns of PR (proteorhodopsin) and DUF2237 have the same appearance, and it can be estimated that these two genes have some relationship. The figure on the right is a bar graph that examines the phototaxis of microorganisms with DUF2237 and ones without it. ⊿DUF2237 is a microorganism that does not have DUF2237; WT is a microorganism that has it.
The protein proteorhodopsin, which is said to be possessed by 40% of the microorganisms on the surface of the ocean. When exposed to light, proteorhodopsin produces a substance called ATP, which is a source of energy. In the table on the left in Fig. 2, the patterns of proteorhodopsin and the function-unknown gene DUF2237 have the same appearance, so it can be estimated that DUF2237 may also have a function related to light. Next, when we examined the microorganisms (WT) that are already known to have DUF2237 and the microorganisms from which DUF2237 has been removed (⊿DUF2237) (see the bar graph on the right in Fig. 2), it was found that those microorganisms that have DUF2237 have stronger phototaxis (property of moving toward light) than those who do not have DUF2237. From this evidence, it can be said that the function-unknown gene DUF2237 is also a gene related to light.
Aiming to establish a more accurate method
In order to know the exact correlation between genes, it is necessary to consider points such as (a) whether the species which are being compared are independent of each other; and (b) whether or not there is a spurious correlation between the genes.
Regarding (a) whether the species which are being compared are independent of each other, we used machine learning to estimate the ancestral genome from the genome data of the existing organism species and the phylogenetic tree, and replaced the framework of the species with patterns of gene gain or loss (Fig. 3). Since the estimation is based on the process of evolution, it is possible to remove closely related elements such as siblings. Important features of my method are the ability to cluster genes according to their characteristics, and the ability to vary the speed of evolution from cluster to cluster. As a result, the accuracy of gene gain/loss patterns has improved dramatically.
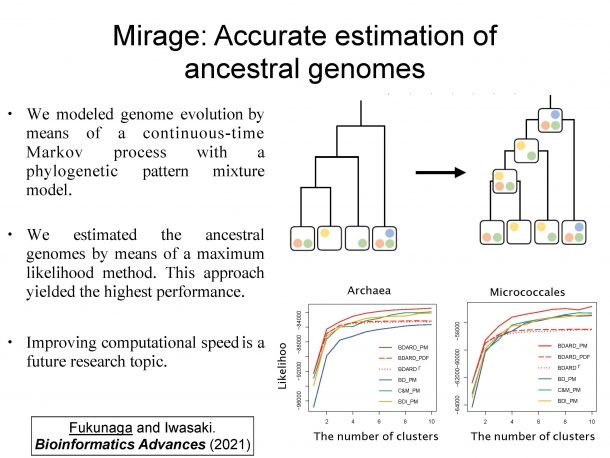
Figure 3. Mirage: Accurate estimation of ancestral genomes
This figure shows the patterns of gene gain and loss using a phylogenetic tree, tracing the ancestral genomes. In the lower right figure, the horizontal axis represents the number of gene clusters and the vertical axis represents accuracy. It can be seen that the accuracy of the pattern increases as the speed of evolution is diversified by increasing the number of clusters.
In the work described above, it is also necessary to estimate the phylogenetic tree itself from the genomes of the extant species. For a more accurate method of estimating the phylogenetic tree, I am conducting research on means of applying machine learning to improve estimation accuracy by applying the phylogenetic tree to the hyperbolic space, which is a space suitable for expressing branching by a vector (quantity with size and direction), rather than the usual Euclidean space.
Regarding (b) whether or not there is a spurious correlation between the genes, I am trying to remove spurious correlations by estimating the original parameters from the genomic data by means of machine learning, using a mathematical model called the Ising Model.
The above is a part of the method that I am working on for estimation of the function-unknown genes of microorganisms. The goal is to develop more accurate software and to make it useful for researchers around the world.
Interview and composition: AIMONO, Keiko
In cooperation with: Waseda University Graduate School of Political Science J-School







